Neuro-Metabolic Homeostasis: The Role of NAD+ in Modulating Neuro-Inflammatory Biomarkers
A deep dive into how NAD+ acts as a critical signaling molecule in the CNS, regulating microglial polarization and suppressing neuro-inflammation through the SIRT1 axis. This article explores the biochemistry of immunometabolism and potential synergies with research peptides like SS-31 and Semax.
Introduction: The Bioenergetic Basis of Neuro-Inflammation
The central nervous system (CNS) is practically voracious in its energy demands, consuming approximately 20% of the body’s total oxygen and glucose despite representing only 2% of total body mass. This intense metabolic activity requires a robust and constant supply of bioenergetic substrates. At the heart of this metabolic engine lies Nicotinamide Adenine Dinucleotide (NAD+), a coenzyme ubiquitous in all living cells. While historically categorized primarily as an electron transporter in redox reactions, contemporary research has elucidated a far more complex role for NAD+: it is a critical signaling molecule governing the transition between homeostasis and inflammation within the brain.
This article explores the mechanisms by which intracellular NAD+ levels dictate the behavior of microglia—the brain's resident immune cells. Current literature suggests that the decline of NAD+ is not merely a symptom of aging or pathology but a driver of "inflammaging," a chronic, low-grade inflammatory state. For researchers investigating neuroprotection, understanding the interplay between NAD+ depletion, mitochondrial dysfunction, and microglial polarization provides a roadmap for utilizing peptides and metabolic agents in CNS studies.
The Biochemistry of NAD+ in the CNS
To understand the neuro-inflammatory role of NAD+, one must first appreciate its metabolic lifecycle. NAD+ exists in an oxidized form (NAD+) and a reduced form (NADH). The ratio of NAD+/NADH is a critical indicator of the cellular metabolic state. In neurons and glial cells, NAD+ is synthesized via three distinct pathways:
- The Precursor Salvage Pathway: The dominant pathway in the mammalian brain, where Nicotinamide (NAM) is converted to Nicotinamide Mononucleotide (NMN) by the rate-limiting enzyme NAMPT, and subsequently to NAD+.
- The De Novo Pathway: De novo synthesis from tryptophan, primarily occurring in the liver but also utilized by specific immune cells, including macrophages and microglia under stress.
- The Preiss-Handler Pathway: Utilization of Nicotinic Acid (NA).
Research indicates that as organisms age, the activity of NAMPT declines while the activity of NAD+-consuming enzymes—specifically PARPs (Poly ADP-ribose polymerases) and CD38—increases. This creates a supply-and-demand deficit. In the context of the CNS, this deficit is catastrophic, leading to a failure in mitochondrial efficiency and the activation of inflammatory cascades.
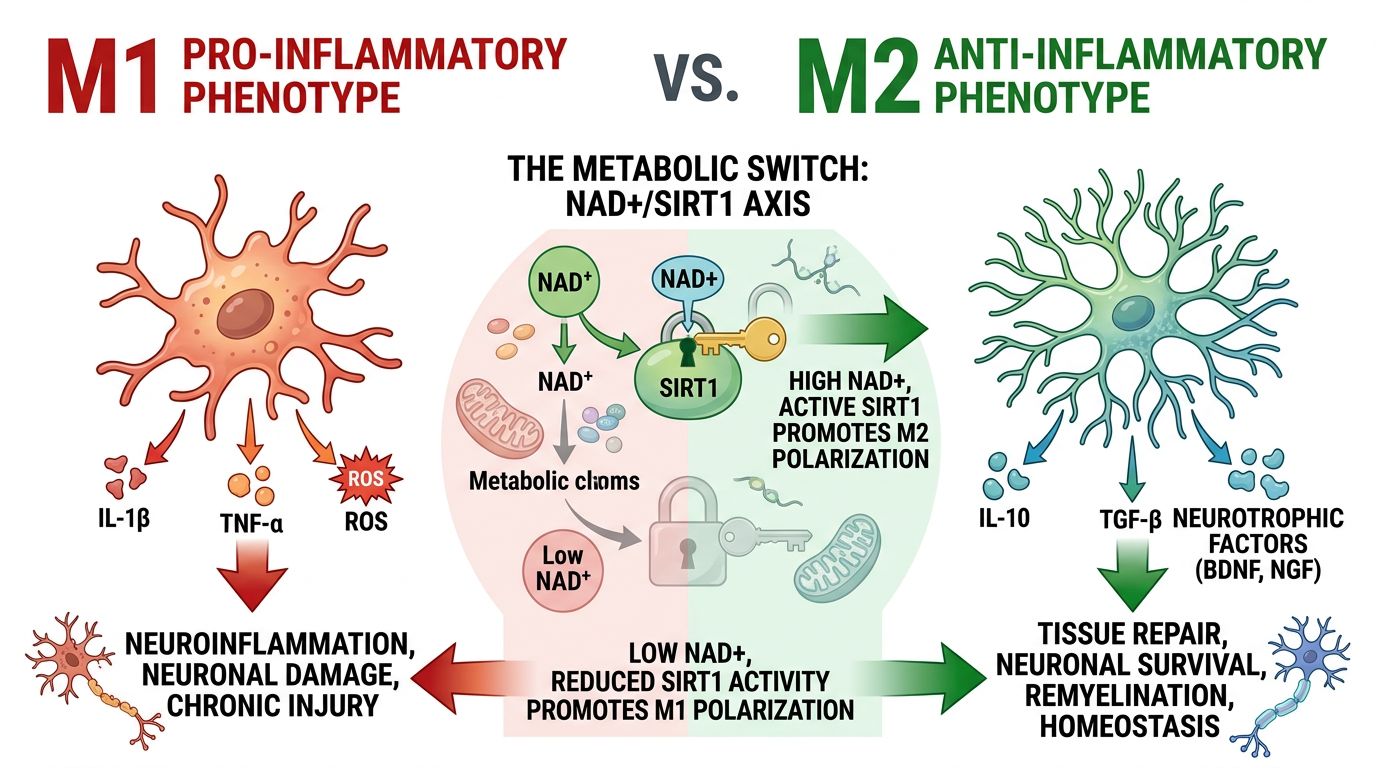
Microglial Polarization: The Metabolic Switch
Microglia serve as the first line of defense in the CNS. They are highly plastic cells that can shift between varying phenotypes, historically simplified into two categories:
- M1 (Pro-inflammatory): Associated with the release of cytokines like TNF-α, IL-1β, and IL-6. This phenotype is neurotoxic if sustained.
- M2 (Anti-inflammatory/Restorative): Associated with tissue repair, phagocytosis of debris (such as amyloid plaques), and release of neurotrophic factors.
Crucially, the switch between M1 and M2 is metabolically regulated. M1 microglia rely on glycolysis (the Warburg effect), allowing for rapid ATP production but resulting in high oxidative stress. M2 microglia rely on oxidative phosphorylation (OXPHOS), which is efficient and sustainable. NAD+ is the linchpin of this switch. Sufficient intracellular NAD+ supports the TCA cycle and OXPHOS, biasing microglia toward the restorative M2 phenotype. Conversely, NAD+ depletion forces cells into glycolysis, locking them into an aggressive M1 inflammatory state.
Table 1: Metabolic Characteristics of Microglial Phenotypes
| Feature | M1 Microglia (Pro-Inflammatory) | M2 Microglia (Anti-Inflammatory) |
|---|---|---|
| Primary Metabolism | Glycolysis | Oxidative Phosphorylation (OXPHOS) |
| NAD+/NADH Ratio | Low | High |
| Mitochondrial Function | Fragmented, low efficiency | Fused, high efficiency |
| Sirtuin Activity | Inhibited (esp. SIRT1) | Active |
| Research Implication | Target for suppression via KPV or Thymosin Alpha-1 | Promote maintenance via NAD+ and SS-31 |
Mechanism of Action: The SIRT1-NF-κB Axis
The protective effects of NAD+ are largely mediated through Sirtuins, a family of NAD+-dependent deacetylases. In the brain, SIRT1 is of paramount importance. SIRT1 monitors cellular energy status (via NAD+ levels) and modulates gene expression accordingly.
Under conditions of high NAD+, SIRT1 is activated and deacetylates the p65 subunit of NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells). NF-κB is the master regulator of the inflammatory response. When deacetylated by SIRT1, NF-κB loses its ability to bind to DNA, thereby preventing the transcription of pro-inflammatory cytokines. This is the molecular mechanism by which high NAD+ levels dampen neuro-inflammation.
When NAD+ levels fall, SIRT1 activity plummets. NF-κB remains acetylated and active, driving the chronic expression of inflammatory markers found in neurodegenerative models. This highlights the potential of using NAD+ alongside peptides like Epithalon, which also influences telomerase and longevity pathways, to maintain genomic stability and reduce inflammatory signaling.
The Role of CD38 and Therapeutic Inhibition
A significant barrier to maintaining NAD+ levels in the aging brain is the enzyme CD38. While CD38 has roles in calcium signaling, it is also a potent NADase (an enzyme that consumes NAD+). Research has shown that in aging microglia, CD38 expression is upregulated by inflammatory cytokines given off by senescent cells (the SASP phenotype).
This creates a vicious cycle: Inflammation increases CD38 -> CD38 depletes NAD+ -> Low NAD+ inhibits SIRT1 -> Inhibited SIRT1 fails to stop Inflammation -> More Inflammation.
Recent investigations into the small molecule 5-amino-1mq focus on its ability to inhibit NNMT, an enzyme involved in adipose metabolism, but parallel research suggests that modulating NAD+ consumption pathways is a viable strategy. By inhibiting enzymes that waste NAD+, researchers aim to restore the pool available for Sirtuin activation and mitochondrial maintenance.
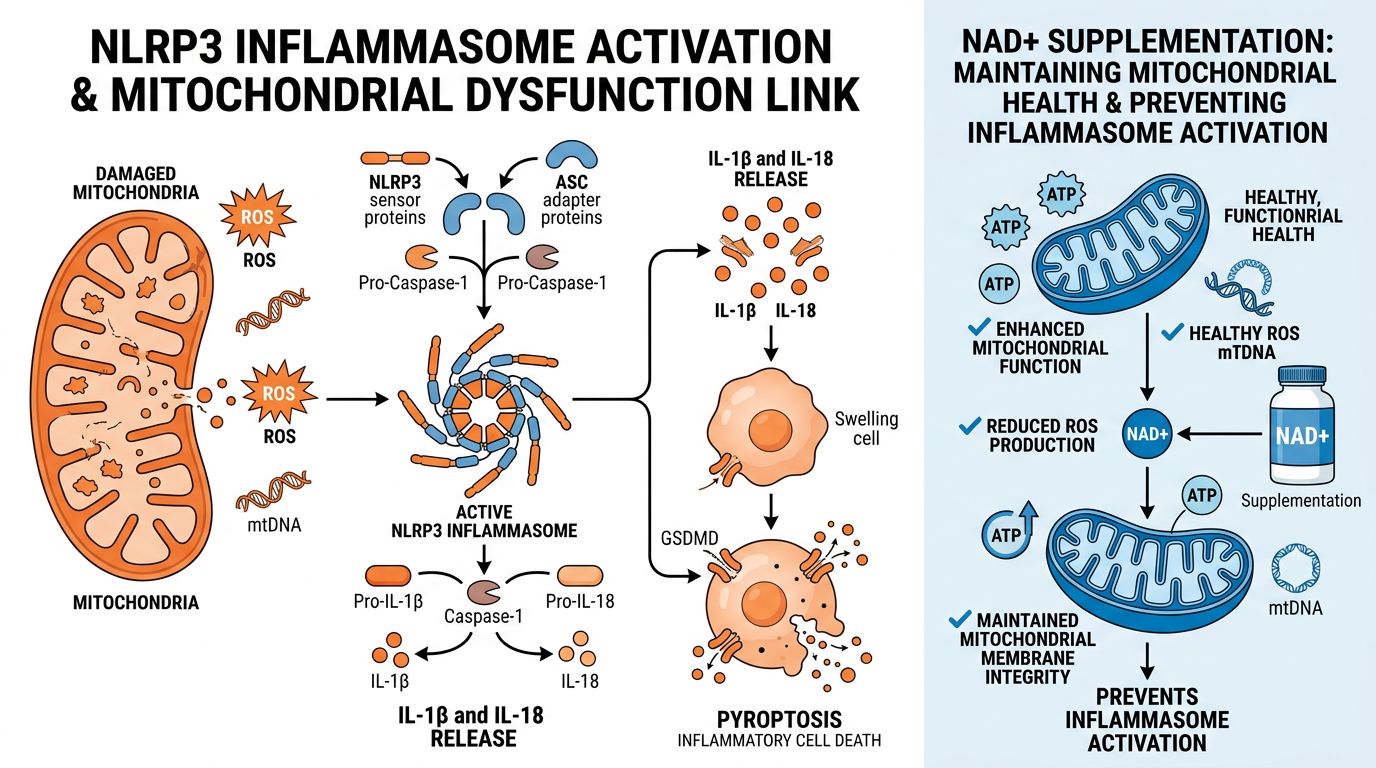
Mitochondrial Dysfunction and the NLRP3 Inflammasome
Beyond the nucleus, NAD+ is vital for mitochondrial integrity. Mitochondria in low-NAD+ environments exhibit increased production of Reactive Oxygen Species (ROS). Mitochondrial ROS (mtROS) acts as a trigger for the NLRP3 inflammasome, a multiprotein complex that processes pro-IL-1β into its active form.
This links NAD+ research directly to mitochondrial peptides. For instance, SS-31 (Elamipretide) targets the inner mitochondrial membrane to stabilize cardiolipin and improve electron transport chain efficiency. When combined with NAD+ replenishment, SS-31 may offer a synergistic approach: NAD+ provides the fuel, while SS-31 tunes the engine, minimizing ROS production and preventing NLRP3 activation. Similarly, MOTS-c, a mitochondrial-derived peptide, assists in metabolic regulation, further supporting the energy homeostasis required to prevent neuro-inflammation.
Synergies in peptide Research: Moving Beyond Monotherapy
In the laboratory setting, the restoration of neuro-metabolic homeostasis often requires a multi-faceted approach. While NAD+ addresses the fundamental energetic deficit, other peptides modulate downstream signaling or structural repair. At Alpha Carbon Labs, we ensure rigorous quality control to support these complex experimental designs.
1. Neurotrophic Support
While NAD+ improves valid metabolic parameters, peptides like Cerebrolysin, Semax, and Selank provide direct neurotrophic support. Semax, for example, modulates BDNF (Brain-Derived Neurotrophic Factor) expression. A study utilizing both NAD+ precursors and Semax could theoretically evaluate if metabolic optimization enhances the efficacy of neurotrophic signaling.
2. Structural Repair
Peptides such as BPC-157 are researched for their angiogenic and reparative properties. In models of Traumatic Brain Injury (TBI) or excitotoxicity, the metabolic crash often impedes repair mechanisms. Ensuring adequate NAD+ availability may be a prerequisite for the reparative processes initiated by BPC-157.
3. Anti-Inflammatory Modulation
For research specifically targeting the inflammatory cascade, Thymosin Alpha-1 and VIP (Vasoactive Intestinal Peptide) are often studied. However, the peptide KPV is gaining traction for its potent anti-inflammatory effects derived from alpha-MSH. Combining KPV's direct cytokine inhibition with NAD+'s upstream metabolic regulation represents a comprehensive strategy for dampening CNS inflammation.
Research Protocols and Stability
When handling NAD+ for research purposes, stability is a primary concern. NAD+ is hygroscopic and sensitive to temperature degradation. Researchers must ensure that substrates are stored in lyophilized forms at -20°C until reconstitution. At Alpha Carbon Labs, our synthesis protocols ensure that the lyophilized NAD+ retains maximum purity, minimizing the presence of degradation byproducts that could interfere with enzymatic assays.
Furthermore, because NAD+ does not easily cross the plasma membrane of all cell types intact (often requiring conversion to NMN or NR extracellularly), delivery vectors and concentrations must be calibrated carefully. Newer research suggests that connexin 43 hemichannels may facilitate direct NAD+ transport in certain glial populations, opening new avenues for administration protocols.
Conclusion
The modulation of neuro-inflammatory biomarkers via NAD+ represents a convergence of immunology and metabolism—immunometabolism. The evidence is compelling: NAD+ is not just a passive energy carrier but an active governor of microglial phenotype. By maintaining high cytosolic and nuclear NAD+ levels, it is possible to keep SIRT1 active, NF-κB suppressed, and mitochondria functional.
For the research community, this underscores the importance of looking beyond receptors and neurotransmitters to the bioenergetic foundation of the cell. Whether investigated in isolation or in synergy with agents like SS-31, Semaglutide (for its metabolic benefits), or CJC-1295, NAD+ remains a critical variable in the equation of neural health and longevity. As we continue to unravel the complexities of the aging brain, high-purity metabolic substrates will remain essential tools for discovery.
References
- 1. Minhas, P. S., et al. (2019). Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nature Immunology, 20, 50–63.
- 2. Verdin, E. (2015). NAD⁺ in aging, metabolism, and neurodegeneration. Science, 350(6265), 1208-1213.
- 3. Gan, L., & Mucke, L. (2008). Paths of neurodegeneration: the peculiar case of autophagy in TBI. Neuron, 58(1), 10-25. (Context: Sirtuins and Neuroprotection).
- 4. Lautrup, S., Sinclair, D. A., Mattson, M. P., & Fang, E. F. (2019). NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metabolism, 30(4), 630-655.
- 5. Camacho-Pereira, J., et al. (2016). CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metabolism, 23(6), 1127-1139.
- 6. Cho, S. H., et al. (2015). SIRT1 deficiency in microglia contributes to amyloid-beta-associated cognitive decline. Aging Cell, 14(6), 994-1003.
- 7. Covarrubias, A. J., et al. (2021). NAD+ metabolism and its roles in cellular processes during aging. Nature Reviews Molecular Cell Biology, 22, 119–141.
- 8. Braidy, N., et al. (2019). NAD+ therapy in age-related degenerative disorders: A benefit/risk analysis. Experimental Gerontology, 115, 20-37.
- 9. Gomes, A. P., et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell, 155(7), 1624-1638.
- 10. Xie, N., et al. (2020). NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduction and Targeted Therapy, 5, 227.
All research information is for educational purposes only. The statements made within this website have not been evaluated by the US Food and Drug Administration. The statements and the products of this company are not intended to diagnose, treat, cure or prevent any disease.