Synergistic Mitigation of Myocellular Decline: The Intersection of NAD+ and ACE-031 Signaling
This article explores the synergistic research potential of combining NAD+ and ACE-031 to mitigate age-related sarcopenia. It examines the physiological mechanisms where metabolic fuel meets myostatin inhibition for optimal myocellular maintenance.
The progressive decline of skeletal muscle mass and function, clinically termed sarcopenia, represents one of the most pervasive hallmarks of biological aging. For researchers in the fields of gerontology and exercise physiology, mitigating this decline is not merely about preserving aesthetics but about maintaining metabolic homeostasis, mobility, and systemic health in aging models. While the etiology of sarcopenia is multifactorial involving neural, hormonal, and environmental components, two primary molecular drivers have emerged as critical targets for intervention: mitochondrial dysfunction stemming from Nicotinamide Adenine Dinucleotide (NAD+) depletion, and the upregulation of negative regulators of muscle mass, specifically the Transforming Growth Factor-beta (TGF-β) superfamily ligands like myostatin.
Recent investigations into peptide therapeutics have begun to explore the intersection of these two distinct pathways. On one hand, we have NAD+, a coenzyme central to cellular metabolism and sirtuin activation. On the other, we have ACE-031 (Acvr2b-Fc), a recombinant fusion protein that acts as a soluble decoy receptor to trap myostatin and activins. This article provides a comprehensive analysis of the potential synergy between these agents, proposing that simultaneous intervention targeting bioenergetic restoration (via NAD+) and the release of anabolic brakes (via ACE-031) may offer a superior modality for reversing myocellular decline than either agent alone.
The Pathophysiology of Sarcopenia: A Dual-Failure Model
To understand the utility of combining NAD+ and ACE-031, one must first dissect the cellular mechanisms underlying sarcopenia. In murine models, age-related muscle loss is characterized by two concurrent failures: the failure of maintenance (catabolism) and the failure of regeneration (anabolism).
Mitochondrial Dysfunction and the NAD+ Crash
Skeletal muscle carries a high energetic demand. Mitochondria within myocytes are responsible for generating the ATP required for contraction and cellular maintenance. As organisms age, systemic levels of NAD+ decline precipitously. NAD+ is a critical substrate for enzymes involved in DNA repair (PARPs) and, more importantly for muscle, the sirtuins (SIRT1 and SIRT3).
Research indicates that when nuclear NAD+ levels drop, a pseudohypoxic state is induced, disrupting nuclear-mitochondrial communication. This leads to a reduction in mitochondrial density and efficiency. Without adequate ATP and sirtuin activity, muscle fibers—particularly type II fast-twitch fibers—become susceptible to oxidative stress and apoptosis, leading to atrophy.
The Myostatin/Activin Brake
Simultaneously, the aging muscle environment often sees an upregulation or increased sensitivity to catabolic signaling. Myostatin (GDF-8) is a potent negative regulator of muscle growth. It binds to the Activin type IIB receptor (ActRIIB) on muscle cell membranes, initiating a signaling cascade via Smad2/3 that inhibits protein synthesis and blocks the proliferation of satellite cells (muscle stem cells).
However, myostatin is not acting alone. Other ligands in the TGF-β superfamily, such as Activin A and GDF11, also signal through ActRIIB to limit muscle growth. In aging models, inflammation often increases Activin A levels, further exacerbating muscle wasting. This creates a "double-brake" system where the body aggressively restricts muscle growth despite external stimuli.
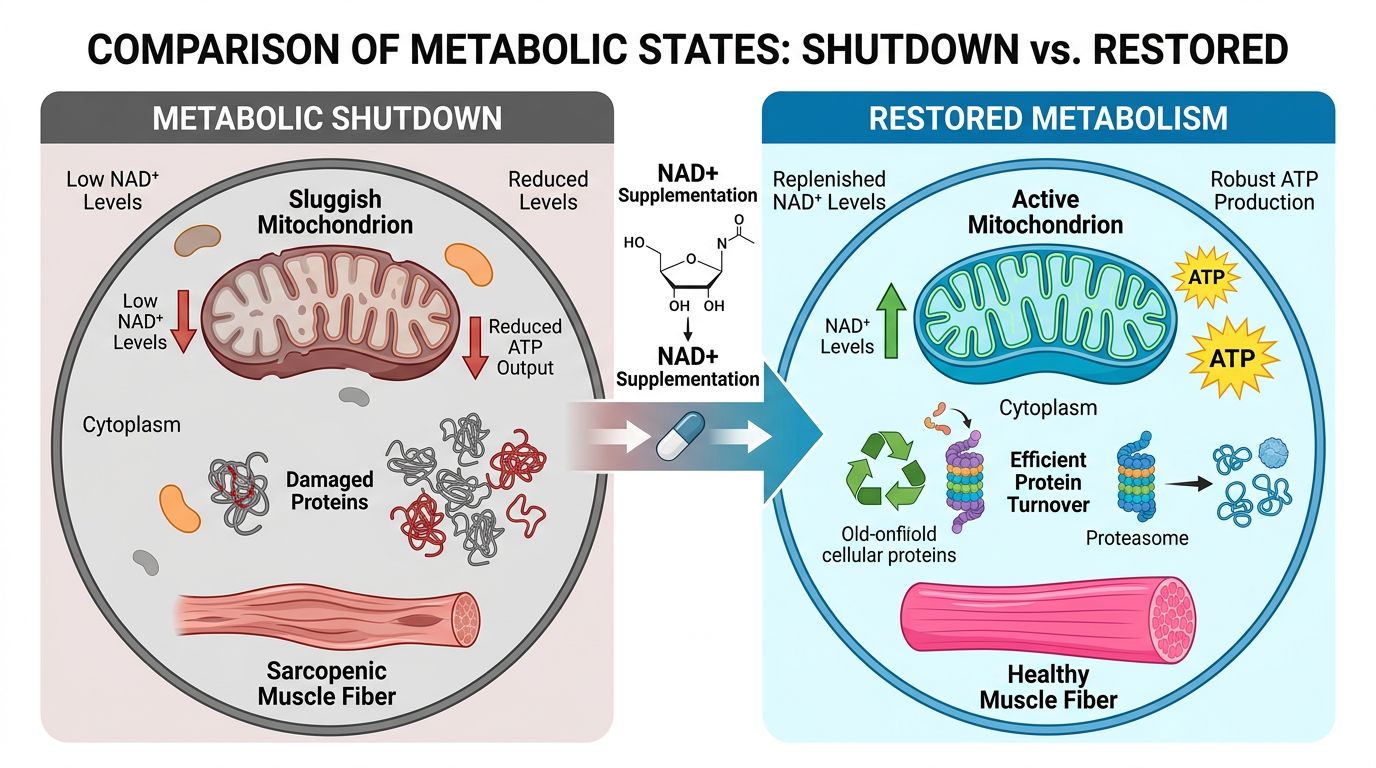
NAD+: The Metabolic Substrate
Exogenous administration of NAD+ in research settings is primarily aimed at restoring the bioenergetic profile of growing or aging tissues to a youthful state. The mechanism of action is foundational.
Mechanism of Action
NAD+ serves as an electron carrier in the Krebs cycle and the Electron Transport Chain (ETC), directly facilitating ATP production. However, its role as a signaling molecule is arguably more critical in the context of sarcopenia. NAD+ is the obligate substrate for Sirtuin 1 (SIRT1), a deacetylase that activates PGC-1α, the master regulator of mitochondrial biogenesis.
By boosting NAD+ levels, researchers have observed:
- Enhanced Mitochondrial Biogenesis: Increased density of healthy mitochondria in skeletal muscle.
- Improved Oxidative Capacity: Shift towards efficient fatty acid oxidation.
- Satellite Cell Activation: SIRT1 is essential for the activation of muscle stem cells upon injury.
- Anti-inflammatory Effects: Reduction in chronic inflammation (inflammaging), which is a key driver of Activin A secretion.
While NAD+ restores the capacity for muscle to function and repair, it does not inherently remove the strong inhibitory signals preventing hypertrophy. It provides the fuel, but the parking brake (myostatin) is still engaged.
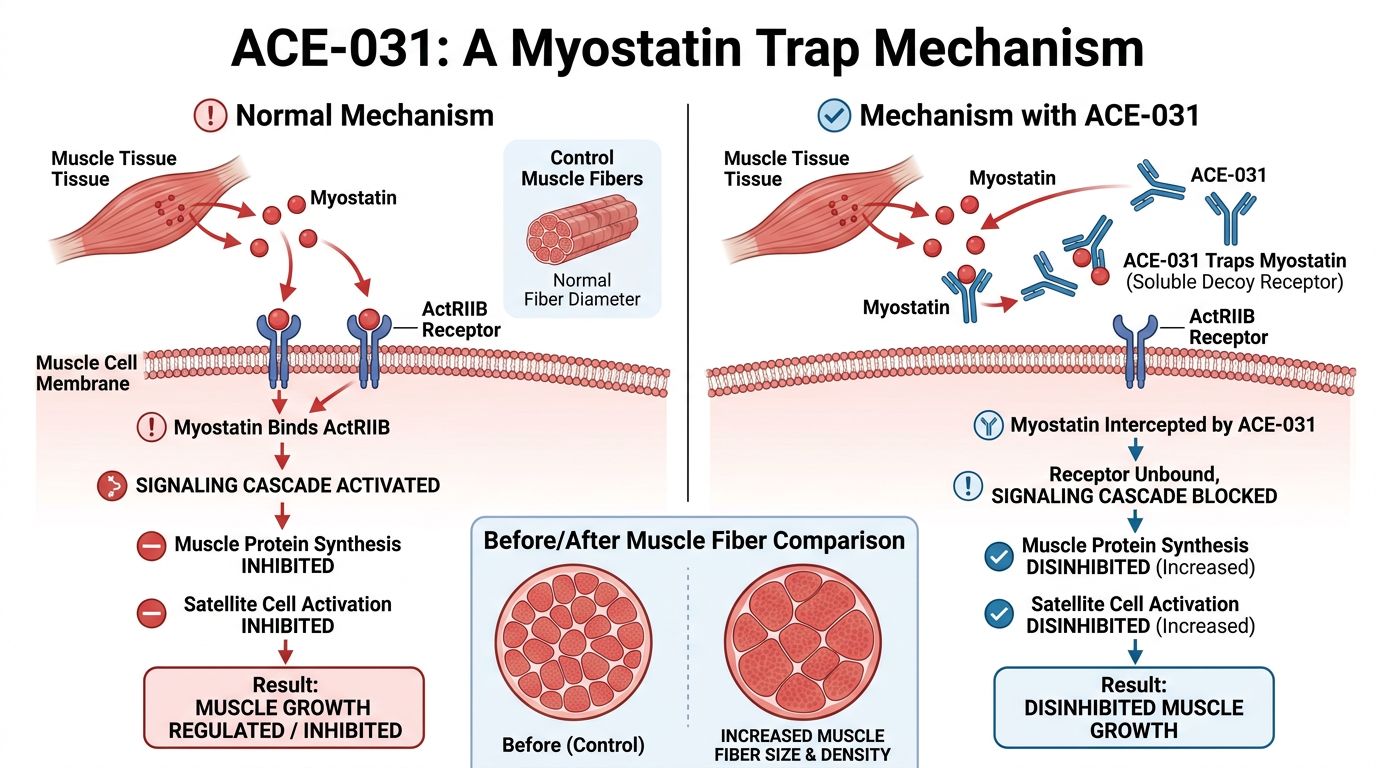
ACE-031: The Soluble Trap
ACE-031 represents a sophisticated approach to myostatin inhibition. Unlike monoclonal antibodies that target only myostatin directly, ACE-031 is a fusion protein consisting of the extracellular domain of the human Activin Receptor Type IIB (ActRIIB) linked to the Fc portion of human IgG1.
Mechanism of Action
ACE-031 acts as a "ligand trap." By circulating in the bloodstream as a soluble receptor, it binds to myostatin, Activin A, Activin B, and GDF11 with high affinity before they can bind to the actual receptors on the muscle cell membrane. This prevents the phosphorylation of Smad2/3 and the subsequent inhibition of the Akt/mTOR pathway.
Research has shown that blocking this specific receptor pathway leads to dramatic increases in lean muscle mass. This occurs through two mechanisms:
- Hypertrophy: Existing muscle fibers increase in cross-sectional area due to upregulated protein synthesis.
- Hyperplasia: Though less common in adults, inhibition of myostatin can facilitate satellite cell proliferation.
Crucially, because ACE-031 binds Activin A and GDF11 in addition to myostatin, it offers a more potent anabolic effect than myostatin-specific antibodies. Activin A is often upregulated in cachectic states, making ACE-031 particularly effective in disease-related biological decline.
The Synergistic Hypothesis: Power and Fuel
The rationale for investigating the combination of NAD+ and ACE-031 stems from a physiological bottleneck observed in aggressive myostatin inhibition therapy. Rapid muscle growth (hypertrophy) induced by agents like ACE-031 imposes a massive metabolic demand on the organism. Protein synthesis is energetically expensive, consuming significant amounts of ATP.
The Metabolic Bottleneck
In aged murine models, where mitochondrial function is already compromised (due to low NAD+), the forced induction of hypertrophy via ACE-031 could potentially stress the cellular machinery. If the mitochondria cannot keep up with the energy demands of rapid growth, the muscle quality might suffer—resulting in large but metabolically inefficient muscle fibers (a phenomenon sometimes seen in glycolytic shifts).
The Cooperative Effect
By co-administering NAD+, researchers aim to "supercharge" the mitochondria to support the rapid tissue accretion driven by ACE-031. The hypothesis dictates that:
- ACE-031 removes the biochemical brakes, allowing for unchecked protein synthesis and satellite cell recruitment.
- NAD+ ensures that the mitochondrial network expands concurrently with the muscle fiber size, preventing metabolic stress and ensuring the new tissue is functional and fatigue-resistant.
This "Power and Fuel" dynamic addresses both the anabolic signaling deficit and the bioenergetic deficit of aging muscle.
Experimental Findings and Molecular Pathways
While direct studies administering this specific combination are emerging, overlapping data from separate trials paints a compelling picture of the mechanisms at play.
mTORC1 vs. AMPK/Sirtuins
Biochemically, there is a delicate balance to be struck. Myostatin inhibition via ACE-031 activates mTORC1, the primary driver of cell growth. NAD+ activates SIRT1 and AMPK, which traditionally dampen mTORC1 to conserve energy during starvation. However, in a nutrient-rich environment (or with sufficient ATP), SIRT1 can actually support muscle growth by keeping the cellular machinery clean (autophagy) and efficient.
Research suggests that when myostatin is inhibited, the subsequent growth requires elevated mitochondrial biogenesis. If mitochondrial regulators (like PGC-1α, activated by NAD+/SIRT1) are not sufficient, the muscle may become large but weak. The addition of NAD+ ensures that PGC-1α activity acts in concert with the mTORC1 drive, creating a phenotype of "oxidative hypertrophy"—muscles that are both big and enduring.
Impact on Bone and Fat
Interestingly, the synergy may extend beyond muscle. ACE-031 has been shown to increase bone mineral density (as the ActRIIB ligands also regulate bone remodeling). NAD+ is crucial for osteoblast function. Furthermore, ACE-031 promotes adiponectin secretion and fat loss. NAD+ improves insulin sensitivity. The combination creates a highly favorable body composition profile in treated subjects.
| Feature | NAD+ Alone | ACE-031 Alone | Combination (NAD+ & ACE-031) |
|---|---|---|---|
| Primary Mechanism | Mitochondrial restoration, Sirtuin activation | Ligand trap for Myostatin/Activins | Anabolic drive + Bioenergetic support |
| Effect on Muscle Mass | Maintenance / Mild prev. of atrophy | Significant Hypertrophy | Supported Hypertrophy (Functional) |
| Mitochondrial Density | Increased | Variable / potentially diluted | Optimized relative to fiber size |
| Fatigue Resistance | High | Neutral | High (Oxidative Hypertrophy) |
Comparative Analysis: ACE-031 vs. Other Agents
When designing research protocols for sarcopenia, researchers often choose between various peptides. Understanding why ACE-031 and NAD+ are chosen over alternatives is vital.
ACE-031 vs. Follistatin
Follistatin is a natural antagonist of myostatin. However, follistatin interacts with a wider range of TGF-β ligands and has a short half-life unless modified. ACE-031 (the soluble receptor fusion) generally offers a more stable and specific pharmacokinetic profile for systemic inhibition in research models. While Follistatin gene therapy is promising, ACE-031 peptide administration allows for precise dose titration and reversibility.
NAD+ vs. SS-31
SS-31 (Elamipretide) is a mitochondrial-targeted peptide that stabilizes cardiolipin but does not necessarily fuel the enzymatic NAD+ pool. While SS-31 is excellent for repairing damaged mitochondria, NAD+ is required for the systemic activation of sirtuins. For a comprehensive anti-aging protocol involving gene expression changes (via sirtuins), NAD+ is often the primary choice, though SS-31 makes an excellent adjunct.
Experimental Dosing and Safety Considerations
Note: All peptides discussed are for research purposes only and not for human consumption. The following discusses data from animal studies.
Dosing Protocols in Literature
In murine studies involving ACE-031, dosages typically range from 10 mg/kg administered once or twice weekly via subcutaneous injection. The effects on muscle mass are often noted within 2-4 weeks. NAD+ protocols vary significantly, but frequent administration (daily or alternate days) is common due to the rapid consumption of the coenzyme. Typical murine doses can range from 100-300 mg/kg intraperitoneally or subcutaneously.
When combining, researchers must monitor for potential side effects associated with rapid tissue growth, such as tendon stress. Interestingly, some data suggests that because ACE-031 improves bone density, it may offer better structural support for larger muscles than myostatin knockout genetics alone.
Safety Profile and Quality Control
In early human clinical trials for Duchenne Muscular Dystrophy, ACE-031 showed promise but trials were paused due to minor adverse events involving gum bleeding and epistaxis (nosebleeds). It is hypothesized that because the ActRIIB receptor is also involved in vascular regulation (ALK1 pathway crosstalk), high systemic doses caused telangiectasia. In research settings, this necessitates careful observation of angiogenic markers.
For any research involving such potent signaling modifiers, the purity of the peptide is paramount. Impurities in longer fusion proteins like ACE-031 can lead to immunogenic responses in test subjects, confounding data. It is essential to source materials that have undergone rigorous HPLC and Mass Spectrometry analysis. Researchers should always verify COA documents and understand the peptide synthesis process used.
Advanced Combinations: The "Golden Quartet"
For advanced research looking at total musculoskeletal rejuvenation, the NAD+ and ACE-031 stack can be theoretically expanded. The following agents act on complementary pathways:
- BPC-157: Known as the "Body Protection Compound," this pentadecapeptide accelerates the healing of tendons and ligaments. Given that ACE-031 rapidly strengthens muscle, the connective tissue must adapt. BPC-157 can assist in collagen organization at the myotendinous junction.
- MK-677 (Ibutamoren): While ACE-031 releases the brake, MK-677 pushes the accelerator by stimulating the release of Growth Hormone (GH) and IGF-1. The combination of myostatin inhibition (ACE-031) and IGF-1 elevation (MK-677) provides a vertical and horizontal attack on muscle anabolism.
- MOTS-c: A mitochondrial-derived peptide that mimics the benefits of exercise. Using MOTS-c alongside NAD+ provides a profound metabolic signal that mimics endurance training, potentially guiding the ACE-031-induced muscle towards a functional, athletic phenotype rather than purely cosmetic bulk.
Future Directions
The intersection of metabolic therapeutics and hypertrophic agents represents the frontier of longevity research. As our understanding of sarcopenia evolves from a simple "loss of mass" to a complex "bioenergetic failure," the tools we use must evolve concurrently. ACE-031 provides the mechanical possibility of regrowing lost tissue, while NAD+ provides the thermodynamic reality to sustain it.
Current research efforts are focused on optimizing the ratio of these agents to minimize off-target vascular effects while maximizing musculoskeletal integrity. The ability to decouple muscle growth from the constraints of age-related metabolic decline offers hope for extending the healthspan of model organisms, with significant implications for future therapeutic strategies.
At Alpha Carbon Labs, we are committed to providing the highest purity research peptides to facilitate this critical investigation. For researchers aiming to push the boundaries of myocellular science, the combination of NAD+ and ACE-031 remains one of the most promising avenues for exploration.
References
- 1. Attie, K. M., et al. (2013). A single ascending-dose study of muscle growth factor trap ACE-031 in healthy postmenopausal women. Muscle & Nerve, 47(3), 416–423.
- 2. Gomes, A. P., et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell, 155(7), 1624–1638.
- 3. Lee, S. J., & McPherron, A. C. (2001). Regulation of myostatin activity and muscle growth. Proceedings of the National Academy of Sciences, 98(16), 9306–9311.
- 4. Campbell, C., et al. (2017). Myostatin inhibition with ACE-031 in boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle & Nerve, 55(4), 458-464.
- 5. Cantó, C., et al. (2015). NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metabolism, 22(1), 31-53.
- 6. Zhou, X., et al. (2010). Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell, 142(4), 531-543.
- 7. Imai, S., & Guarente, L. (2014). NAD+ and sirtuins in aging and disease. Trends in Cell Biology, 24(8), 464-471.
- 8. Cadena, S. M., et al. (2010). Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of IGF-1 signaling. Journal of Applied Physiology, 109(3), 635-642.
- 9. Faitg, J., et al. (2020). Crosstalk between mitochondrial biogenesis and control of muscle mass in sarcopenia. Cells, 9(5), 1251.
- 10. Latres, E., et al. (2017). Myostatin blockade with a fully human monoclonal antibody induces muscle hypertrophy and reverses muscle atrophy in young and aged mice. Skeletal Muscle, 5, 34.
All research information is for educational purposes only. The statements made within this website have not been evaluated by the US Food and Drug Administration. The statements and the products of this company are not intended to diagnose, treat, cure or prevent any disease.